Gene PCA DEMO LAB#
## NOTE: This is Python 3 code.
import pandas as pd
import numpy as np
import random as rd
from sklearn.decomposition import PCA
from sklearn import preprocessing
from sklearn.preprocessing import StandardScaler
import matplotlib.pyplot as plt
Data Generation Code#
## In this example, the data is in a data frame called data.
## Columns are individual samples (i.e. cells)
## Rows are measurements taken for all the samples (i.e. genes)
## Just for the sake of the example, we'll use made up data...
genes = ['gene' + str(i) for i in range(1,101)]
wt = ['wt' + str(i) for i in range(1,6)]
ko = ['ko' + str(i) for i in range(1,6)]
data = pd.DataFrame(columns=[*wt, *ko], index=genes)
for gene in data.index:
data.loc[gene,'wt1':'wt5'] = np.random.poisson(lam=rd.randrange(10,1000), size=5)
data.loc[gene,'ko1':'ko5'] = np.random.poisson(lam=rd.randrange(10,1000), size=5)
print(data.head())
print(data.shape)
wt1 wt2 wt3 wt4 wt5 ko1 ko2 ko3 ko4 ko5
gene1 652 663 703 645 646 745 767 768 774 818
gene2 122 115 123 113 117 905 875 925 980 884
gene3 614 610 623 601 608 780 765 729 729 692
gene4 24 26 16 27 32 444 389 409 375 410
gene5 700 725 695 647 706 937 924 898 891 964
(100, 10)
Perform PCA on the data#
# First center and scale the data
scaled_data = preprocessing.scale(data.T) ## scale need the samples to be rows!
## better method:
# StandardScaler().fit_transform(data.T)
pca = PCA() # create a PCA object
pca.fit(scaled_data) # do the math
pca_data = pca.transform(scaled_data) # get PCA coordinates for scaled_data
Draw a scree plot and a PCA plot#
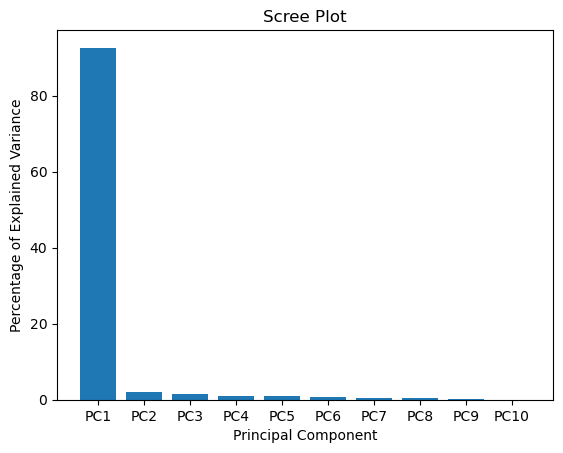
#The following code constructs the Scree plot
per_var = np.round(pca.explained_variance_ratio_* 100, decimals=1)
labels = ['PC' + str(x) for x in range(1, len(per_var)+1)]
plt.bar(x=range(1,len(per_var)+1), height=per_var, tick_label=labels)
plt.ylabel('Percentage of Explained Variance')
plt.xlabel('Principal Component')
plt.title('Scree Plot')
plt.show()
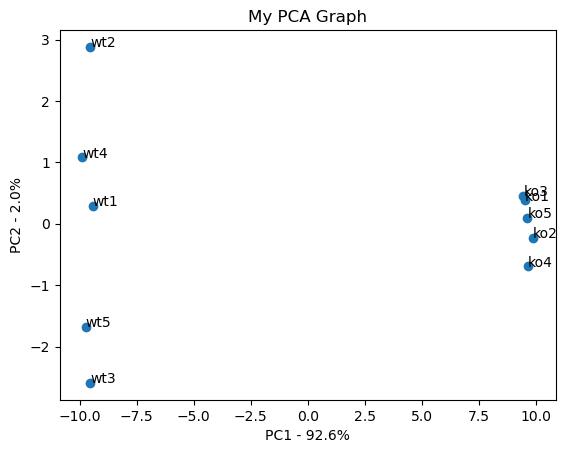
#the following code makes a fancy looking plot using PC1 and PC2
pca_df = pd.DataFrame(pca_data, index=[*wt, *ko], columns=labels)
plt.scatter(pca_df.PC1, pca_df.PC2)
plt.title('My PCA Graph')
plt.xlabel('PC1 - {0}%'.format(per_var[0]))
plt.ylabel('PC2 - {0}%'.format(per_var[1]))
for sample in pca_df.index:
plt.annotate(sample, (pca_df.PC1.loc[sample], pca_df.PC2.loc[sample]))
plt.show()
Determine which genes had the biggest influence on PC1#
## get the name of the top 10 measurements (genes) that contribute
## most to pc1.
## first, get the loading scores
loading_scores = pd.Series(pca.components_[0], index=genes) ### pc1=index0
## now sort the loading scores based on their magnitude
sorted_loading_scores = loading_scores.abs().sort_values(ascending=False)
# get the names of the top 10 genes
top_10_genes = sorted_loading_scores[0:10].index.values
## print the gene names and their scores (and +/- sign)
print(loading_scores[top_10_genes])
gene83 -0.103892
gene43 -0.103856
gene77 -0.103836
gene70 0.103821
gene29 -0.103809
gene78 0.103802
gene10 0.103797
gene35 0.103796
gene97 0.103790
gene23 -0.103785
dtype: float64